Marfan syndrome
Overview
Marfan syndrome is an inherited disorder that affects connective tissue — the fibers that support and anchor your organs and other structures in your body. Marfan syndrome most commonly affects the heart, eyes, blood vessels and skeleton.
People with Marfan syndrome are usually tall and thin with unusually long arms, legs, fingers and toes. The damage caused by Marfan syndrome can be mild or severe. If your aorta — the large blood vessel that carries blood from your heart to the rest of your body — is affected, the condition can become life-threatening.
Treatment usually includes medications to keep your blood pressure low to reduce the strain on your aorta. Regular monitoring to check for damage progression is vital. Many people with Marfan syndrome eventually require preventive surgery to repair the aorta.
Symptoms
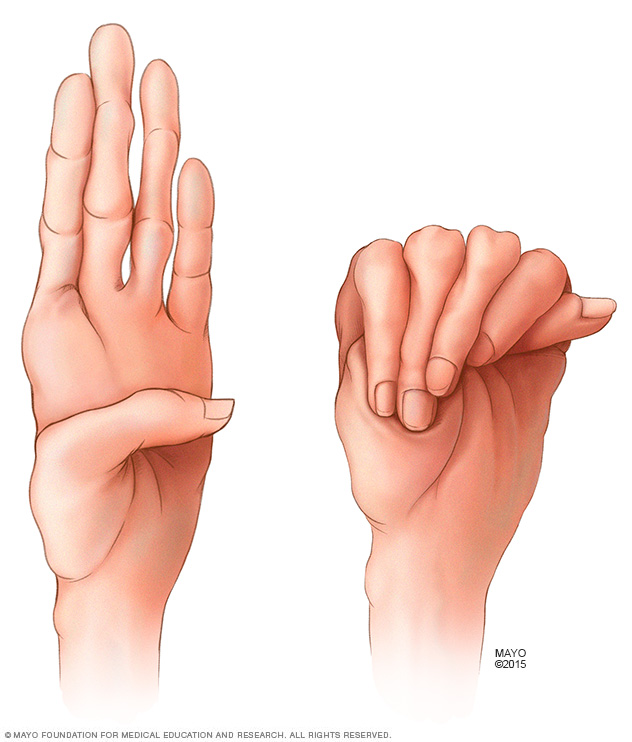
People who have Marfan syndrome typically have especially long fingers. It's common for their thumbs to extend far beyond the edge of their hands when they make a fist.
The signs and symptoms of Marfan syndrome can vary greatly, even among members of the same family, because the disorder can affect so many different areas of the body. Some people experience only mild effects, but others develop life-threatening complications.
Marfan syndrome features may include:
- Tall and slender build
- Disproportionately long arms, legs and fingers
- A breastbone that protrudes outward or dips inward
- A high, arched palate and crowded teeth
- Heart murmurs
- Extreme nearsightedness
- An abnormally curved spine
- Flat feet
When to see a doctor
If you think that you or your child may have Marfan syndrome, talk to your doctor or pediatrician. If your doctor suspects a problem, you'll likely be referred to a specialist for further evaluation.
Causes
Marfan syndrome is caused by a defect in the gene that enables your body to produce a protein that helps give connective tissue its elasticity and strength.
Most people with Marfan syndrome inherit the abnormal gene from a parent who has the disorder. Each child of an affected parent has a 50-50 chance of inheriting the defective gene. In about 25% of the people who have Marfan syndrome, the abnormal gene comes from neither parent. In these cases, a new mutation develops spontaneously.
Risk factors
Marfan syndrome affects men and women equally and occurs among all races and ethnic groups. Because it's a genetic condition, the greatest risk factor for Marfan syndrome is having a parent with the disorder.
Complications
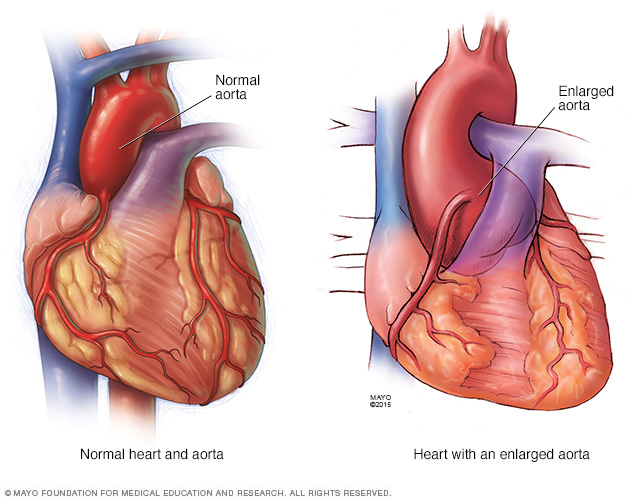
The pressure of blood leaving your heart can cause the wall of your aorta to bulge out, like a weak spot in a tire. In people who have Marfan syndrome, this is most likely to happen at the aortic root — where the artery leaves your heart.
Because Marfan syndrome can affect almost any part of your body, it may cause a wide variety of complications.
Cardiovascular complications
The most dangerous complications of Marfan syndrome involve the heart and blood vessels. Faulty connective tissue can weaken the aorta — the large artery that arises from the heart and supplies blood to the body.
- Aortic aneurysm. The pressure of blood leaving your heart can cause the wall of your aorta to bulge out, like a weak spot in a tire. In people who have Marfan syndrome, this is most likely to happen at the aortic root — where the artery leaves your heart.
- Aortic dissection. The wall of the aorta is made up of layers. Dissection occurs when a small tear in the innermost layer of the aorta's wall allows blood to squeeze between the inner and outer layers of the wall. This can cause severe pain in the chest or back. An aortic dissection weakens the vessel's structure and can result in a rupture, which may be fatal.
- Valve malformations. People who have Marfan syndrome can have weak tissue in their heart valves. This can produce stretching of the valve tissue and abnormal valve function. When heart valves don't work properly, your heart often has to work harder to compensate. This can eventually lead to heart failure.
Eye complications
Eye complications may include:
- Lens dislocation. The focusing lens within your eye can move out of place if its supporting structures weaken. The medical term for this problem is ectopia lentis, and it occurs in more than half the people who have Marfan syndrome.
- Retinal problems. Marfan syndrome also increases the risk of a detachment or tear in the retina, the light-sensitive tissue that lines the back wall of your eye.
- Early-onset glaucoma or cataracts. People who have Marfan syndrome tend to develop these eye problems at a younger age. Glaucoma causes the pressure within the eye to increase, which can damage the optic nerve. Cataracts are cloudy areas in the eye's normally clear lens.
Skeletal complications
Marfan syndrome increases the risk of abnormal curves in the spine, such as scoliosis. It can also interfere with the normal development of the ribs, which can cause the breastbone to either protrude or appear sunken into the chest. Foot pain and low back pain are common with Marfan syndrome.
Complications of pregnancy
Marfan syndrome can weaken the walls of the aorta, the main artery that leaves the heart. During pregnancy, the heart pumps more blood than usual. This can put extra stress on the aorta, which increases the risk of a deadly dissection or rupture.
Diagnosis
Marfan syndrome can be challenging for doctors to diagnose because many connective tissue disorders have similar signs and symptoms. Even among members of the same family, the signs and symptoms of Marfan syndrome vary widely — both in their features and in their severity.
Certain combinations of symptoms and family history must be present to confirm a diagnosis of Marfan syndrome. In some cases, a person may have some features of Marfan syndrome, but not enough of them to be diagnosed with the disorder.
Heart tests
If your doctor suspects Marfan syndrome, one of the first tests he or she may recommend is an echocardiogram. This test uses sound waves to capture real-time images of your heart in motion. It checks the condition of your heart valves and the size of your aorta. Other heart-imaging options include computerized tomography (CT) scans and magnetic resonance imaging (MRI).
If you are diagnosed with Marfan syndrome, you'll need to have regular imaging tests to monitor the size and condition of your aorta.
Eye tests
Eye exams that may be needed include:
- Slit-lamp exam. This test checks for lens dislocation, cataracts or a detached retina. Your eyes will need to be completely dilated with drops for this exam.
- Eye pressure test. To check for glaucoma, your eye doctor may measure the pressure inside your eyeball by touching it with a special tool. Numbing eyedrops are usually used before this test.
Genetic testing
Genetic testing is often used to confirm the diagnosis of Marfan syndrome. If a Marfan mutation is found, family members can be tested to see if they are also affected. You may want to talk to a genetic counselor before starting a family, to see what your chances are of passing on Marfan syndrome to your future children.
Treatment
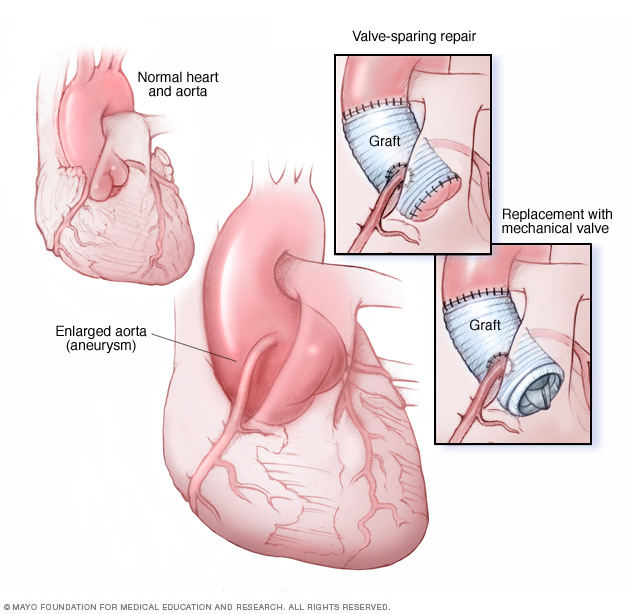
An ascending aortic root aneurysm procedure may be done in two ways. In aortic root replacement, your surgeon removes a section of your aorta and your aortic valve. The aortic section is replaced with an artificial tube (graft). The aortic valve is replaced with a mechanical or biological valve (bottom right image). Some people have valve-sparing aortic root repair (top right image), in which the aortic valve remains in place.
While there is no cure for Marfan syndrome, treatment focuses on preventing the various complications of the disease. To accomplish this, you'll need to be checked regularly for signs that the damage caused by the disease is progressing.
In the past, people who had Marfan syndrome often died young. With regular monitoring and modern treatment, most people with Marfan syndrome can now expect to live a more normal life span.
Medications
Doctors often prescribe blood pressure lowering drugs to help prevent the aorta from enlarging and to reduce the risk of dissection and rupture.
Therapy
The vision problems associated with a dislocated lens in your eye often can be corrected with glasses or contact lenses.
Surgical and other procedures
Depending on your signs and symptoms, procedures might include:
- Aortic repair. If your aorta's diameter reaches about 2 inches (50 millimeters) or if it enlarges rapidly, your doctor may recommend an operation to replace a portion of your aorta with a tube made of synthetic material. This can help prevent a life-threatening rupture. Your aortic valve may need to be replaced as well.
- Scoliosis treatment. When there is significant scoliosis, a consultation with a spine expert is necessary. Bracing and surgery are needed in some cases.
- Breastbone corrections. Surgical options are available to correct the appearance of a sunken or protruding breastbone. Because these operations are often considered to be for cosmetic purposes, your insurance might not cover the costs.
- Eye surgeries. If parts of your retina have torn or come loose from the back of your eye, surgical repair is usually successful. If you have cataracts, your clouded lens can be replaced with an artificial lens.
Lifestyle and home remedies
You may need to avoid competitive sports and certain recreational activities if you're at increased risk of aortic dissection or rupture. Increases in blood pressure, common in activities such as weightlifting, place extra strain on the aorta. Less intense activities — such as brisk walking, bowling, doubles tennis or golf — are generally safer.
Coping and support
Living with a genetic disorder can be extremely difficult for both adults and children. Adults may wonder how the disease will affect their careers, their relationships and their sense of themselves. And they may worry about passing the defective gene to their children.
But Marfan syndrome can be even harder on young people, especially because the often-inherent self-consciousness of childhood and adolescence may be exacerbated by the disease's effect on appearance, academic performance and motor skills.
Helping children cope
Working together, parents, teachers and medical professionals can provide children with both emotional support and practical solutions for some of the more distressing aspects of the disease. For example, children with Marfan syndrome may struggle in school because of vision problems that can be corrected with glasses or contact lenses.
For most young people, cosmetic concerns are at least as important as academic ones. Parents can help by anticipating these concerns and offering solutions, such as:
- Contact lenses instead of glasses
- A brace for scoliosis
- Dental work for crowded teeth
- Clothes that flatter a tall, thin frame
Support groups
People who have Marfan syndrome often find it helpful to talk with others facing the similar challenges. The Marfan Foundation provides a variety of support services online.
Preparing for an appointment
Marfan syndrome can affect many different parts of your body, so you may need to see a variety of medical specialists, such as:
- A cardiologist, a doctor who specializes in heart and blood vessel disorders
- An ophthalmologist, a doctor who specializes in eye disorders
- An orthopedist, a doctor who specializes in structural problems of the skeleton
- A geneticist, a doctor who specializes in genetic disorders
To make the best use of appointment time, plan ahead and have important information available, including:
- Detailed descriptions of all your symptoms
- Details of your past medical history, including any previous surgeries
- Past X-rays and echocardiogram reports, which often can be sent electronically
- A list of all your medications and supplements
What to expect from your doctor
All your doctors will want to hear about your specific symptoms, and whether anyone in your family has had Marfan syndrome or experienced an early, unexplained heart-related disability or death.
Content Last Updated: May 19, 2021
Content provided by Mayo Clinic ©1998-2026 Mayo Foundation for Medical Education and Research (MFMER). All rights reserved. Terms of Use