Congenital adrenal hyperplasia
Overview
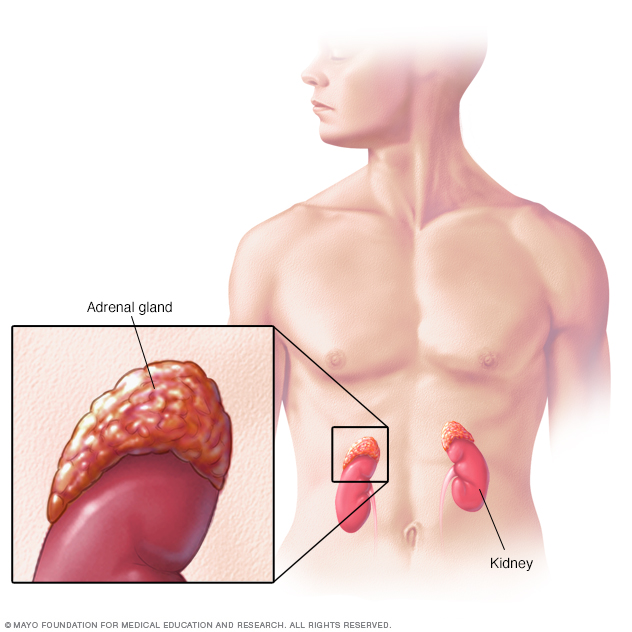
Perched atop each of your kidneys, your adrenal glands produce hormones that help regulate your metabolism, immune system, blood pressure and other essential functions.
Congenital adrenal hyperplasia (CAH) refers to a group of genetic disorders that affect the adrenal glands, a pair of walnut-sized organs above the kidneys. The adrenal glands produce important hormones, including:
- Cortisol, which regulates the body's response to illness or stress
- Mineralocorticoids, such as aldosterone, which regulate sodium and potassium levels
- Androgens, such as testosterone, which are male sex hormones
In people who have CAH, a genetic problem results in a lack of one of the enzymes needed to make these hormones.
Although there is no cure, with proper treatment, most people who have congenital adrenal hyperplasia can lead normal lives.
There are two major types of congenital adrenal hyperplasia:
- Classic CAH. This form is rarer and is usually detected in infancy. Approximately two-thirds of people who have classic CAH have what's known as the salt-losing form, while one-third have what's referred to as the simple-virilizing form.
- Nonclassic CAH. This form is milder and more common, and may not become evident until childhood or early adulthood.
Symptoms
Signs and symptoms of CAH vary, depending on which gene is defective and the level of enzyme deficiency.
Classic CAH
Female infants who have classic CAH may have a condition known as ambiguous genitalia, in which the clitoris is enlarged or the genitals look more like those of a male child. Male infants who have classic CAH have normal appearing genitals. Both male and female infants can be seriously affected by a lack of cortisol, aldosterone or both. This is known as an adrenal crisis, and it can be life-threatening.
The salt-losing form and simple-virilizing form of classic CAH cause children's bodies to produce an insufficient amount of cortisol. These children can have problems maintaining normal blood pressure, normal blood sugar and energy levels, and are more vulnerable to stress. An excess of the male sex hormones can result in short height and early puberty for both boys and girls.
Signs and symptoms of classic CAH in children and adults include:
- Appearance of pubic hair at a very early age
- Rapid growth during childhood, but shorter than average final height
Nonclassic CAH
Often there are no symptoms of nonclassic CAH when a baby is born. The condition is not identified on routine infant blood screening and usually becomes evident in late childhood or early adulthood. Cortisol may be the only hormone that's deficient.
Teenage and adult females who have nonclassic CAH may have normal appearing genitals at birth, but later in life, they may experience:
- Irregular or absent menstrual periods
- Masculine characteristics such as facial hair, excessive body hair and a deepening voice
- Severe acne
In both females and males, signs of nonclassic CAH may also include:
- Early appearance of pubic hair
- Rapid growth during childhood, an advanced bone age and shorter predicted final height
When to see a doctor
Classic CAH is usually detected at birth through required newborn screening or when female babies have ambiguous genitalia. CAH may also be identified when male or female babies show signs of severe illness due to low levels of cortisol, aldosterone or both.
In children who have nonclassic CAH, signs and symptoms of early puberty may appear. If you have concerns about your child's growth or development, make an appointment with your child's doctor.
If you are pregnant and may be at risk of CAH because of your own medical history or your ethnicity, ask your doctor about genetic counseling.
Causes
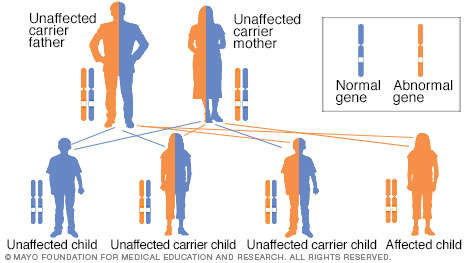
To have an autosomal recessive disorder, you inherit two mutated genes, one from each parent. These disorders are usually passed on by two carriers. Their health is rarely affected, but they have one mutated gene (recessive gene) and one normal gene (dominant gene) for the condition. Two carriers have a 25% chance of having an unaffected child with two normal genes (left), a 50% chance of having an unaffected child who also is a carrier (middle), and a 25% chance of having an affected child with two recessive genes (right).
The most common cause of CAH is the lack of the enzyme known as 21-hydroxylase. CAH may sometimes be called 21-hydroxylase deficiency. There are other much rarer enzyme deficiencies that also cause CAH.
Children who have the condition have two parents who either have CAH themselves or who are both carriers of the genetic mutation that causes the condition. This is known as the autosomal recessive inheritance pattern.
Risk factors
Factors that increase the risk of having CAH include:
- Parents who both have CAH or are both carriers of the genetic defect for the disorder
- Certain ethnic heritages, such as Ashkenazi Jew, but also Hispanic, Italian, Yugoslav and Yupik Inuit
Complications
People who have classic CAH are at risk of adrenal crisis because they have very low levels of cortisol in the blood. This can cause diarrhea, vomiting, dehydration, low blood sugar levels and shock. Adrenal crisis is a life-threatening medical emergency that requires immediate treatment. Aldosterone also may be low, which leads to dehydration and low sodium and high potassium levels. The nonclassic form of CAH doesn't cause adrenal crisis.
Males and females who have either classic or nonclassic CAH may also experience fertility problems.
Prevention
There is no known way to prevent congenital adrenal hyperplasia. If you're thinking of starting a family and you're at risk of having a child with CAH, your doctor may recommend that you see a genetic counselor.
Diagnosis
CAH may be diagnosed before a baby is born, during childhood or later in life.
Prenatal testing
Tests used to diagnose CAH in fetuses include:
- Amniocentesis. This procedure involves using a needle to withdraw a sample of amniotic fluid from the womb and then examining the cells.
- Chorionic villus sampling. This test involves withdrawing cells from the placenta for examination.
Newborns, infants and children
Doctors recommend that all newborns in the United States be screened for genetic 21-hydroxylase deficiency during the first few days of life. This test identifies the classic form of CAH but doesn't identify the nonclassic form.
Diagnosis of CAH in older children and young adults includes:
- Physical exam. If the doctor suspects CAH based on a physical exam and symptoms, the next step is to confirm the diagnosis with blood and urine tests.
- Blood and urine tests. These tests look for abnormal levels of hormones produced by the adrenal glands.
- Gene testing. In older children and young adults, genetic testing may be needed to diagnose CAH.
- Testing to determine a child's sex. In female infants who have severe ambiguous genitalia, tests can be done to analyze chromosomes to identify genetic sex. Also, pelvic ultrasound can be used to identify the presence of female reproductive structures such as the uterus and ovaries.
Treatment
Your doctor will likely refer your child to a doctor who specializes in childhood hormonal issues (pediatric endocrinologist) for treatment of CAH. The health care team may also include other specialists, such as urologists, psychologists and geneticists.
Medications
The goal of treating CAH with medications is to reduce excess androgen production and replace deficient hormones. People who have the classic form of CAH can successfully manage the condition by taking hormone replacement medications throughout their lives. People who have nonclassic CAH may not require treatment or may need only small doses of corticosteroids.
Medications for CAH are taken on a daily basis. During periods of illness or significant stress, such as surgery, additional medications or higher doses may be needed.
Medications may include:
- Corticosteroids to replace cortisol
- Mineralocorticoids to replace aldosterone to help retain salt and get rid of excess potassium
- Salt supplements to help retain salt
Monitoring the effectiveness of medication includes regularly scheduled:
- Physical exams. The doctor will check your child's growth and development, including monitoring changes in height, weight, blood pressure and bone growth.
- Monitoring for side effects. The doctor will monitor your child for side effects, such as the loss of bone mass and impaired growth, particularly if steroid-type replacement medication doses are high and used long term.
- Blood tests to check hormone levels. It's critical to have regular blood tests to ensure hormone levels are balanced. A child who hasn't yet reached puberty needs enough cortisone to suppress androgens so that he or she can grow to a normal height. For females who have CAH, it's important to suppress androgens to minimize unwanted masculine characteristics. On the other hand, too much cortisone can cause Cushing syndrome.
Reconstructive surgery
In some female infants who have severe ambiguous genitalia as a result of classic CAH, doctors may recommend reconstructive surgery to improve genital function and make them look more feminine.
Surgery may involve reducing the size of the clitoris and reconstructing the vaginal opening. The surgery is typically performed between 2 and 6 months of age. Females who have reconstructive genital surgery may need more cosmetic surgery later in life.
Genital surgery is easier to perform when a child is very young. However, some parents choose to wait for surgery until their child is old enough to understand the risks and choose his or her own gender assignment.
Before making decisions about the best treatment approach for your child, talk with your doctor about these issues. Working together, you and your doctor can make informed choices that will help your child thrive.
Psychological support is important to the emotional health and social adjustment of female children who have genital abnormalities.
Prenatal treatment
Synthetic corticosteroids that cross the placenta to the fetus are controversial and considered experimental. More research is needed to determine the long-term safety and the effect of this treatment on fetal brain development.
Coping and support
Early and steady support from family and health care professionals can help your child have normal self-esteem and a satisfying social life. These approaches may help:
- Include psychological counseling in your child's treatment plan as needed
- Seek help from a mental health professional if you're having trouble coping and to help you develop healthy parenting strategies
Preparing for an appointment
You may start by seeing your family doctor or your child's pediatrician. He or she may refer you to a doctor who specializes in the diagnosis and treatment of conditions related to the adrenal glands (pediatric endocrinologist).
Here's some information to help you prepare for your appointment. Consider taking a family member or friend along for support and to help you remember information.
What you can do
To prepare for your appointment:
- Find out if your child needs to follow any pre-appointment restrictions, such as changing food or liquid intake to get ready for blood and urine tests.
- Make a list of any signs and symptoms your child has been experiencing, and for how long.
- Make a list of your child's key medical information, including recent illnesses, any medical conditions, and the names and dosages of any medications, vitamins, herbs or other supplements.
- Prepare questions you want to ask your doctor.
Some basic questions to ask your doctor may include:
- What is likely causing my child's signs and symptoms?
- Are there other possible causes for these symptoms?
- What kinds of tests does my child need?
- What treatment approach do you recommend?
- What are the expected results of treatment?
- What are the possible side effects of treatment?
- How will you monitor my child's health over time?
- What is my child's risk of long-term complications?
- Do you recommend that my child receive psychological counseling?
- Do you recommend that our family meet with a genetic counselor?
Don't hesitate to ask any other questions during your appointment.
What to expect from your doctor
Your doctor is likely to ask you a number of questions. Be ready to answer them to reserve time to go over points you want to focus on. For example, your doctor may ask:
- What are your child's symptoms?
- When did you first begin noticing these symptoms?
- Has anyone in your family been diagnosed with congenital adrenal hyperplasia? If so, do you know how it was treated?
- Are you planning to have more children?
Content Last Updated: October 14, 2021
Content provided by Mayo Clinic ©1998-2026 Mayo Foundation for Medical Education and Research (MFMER). All rights reserved. Terms of Use